1991 Oxford University Press. Nucleic Acids Research, vol. 19, N° 6.
A simple and rapid method for the preparation of plant genomic DNA for PCR analysis
K.Edwards, C.Johnstone and C.Thompson
Plant Biotechnology Section, ICI Seeds, Jealott's Hill Research Station,
Braeknell, Berks, UK
submitted January 2, 1991
The polymerase chain reaction (PCR) has revolutionised the rapid analysis of mammalian genomic DNA (I). However, PCR is less useful in the analysis of plant DNA due to te difficulties in extracting nucleid acids from limited amounts of plant tissue. We have developed a method for the rapid extraction of small amounts of plant genomic DNA suitable for PCR analysis. The method is applicable te a variety of plant species and has the advantage of not requiring any phenol or chloroform extraction. Thus it is possible te complete an extraction within minutes without handling any hazardous organic solvents. Samples for PCR analysis (usually leaf tissue) are collected using the lid of a sterile Eppendorf tube to pinch out a disc of material into te tube. This ensures uniform sample size and also reduces the possibilities of contamination arising from handling the tissue. DNA is extracted as follows: The tissue is macerated (using disposable grinders from Bel-art Products: Scienceware, Pequannock, NJ. 07440 USA. catalog no 992) in the original Eppendorf tube at room temperature, without buffer, for 15 seconds. 400 µl of extraction buffer (200 mM Tris HCI pH G mN4 NaCI, 25 mM EDTA, 0,5% SDS) is added and the sample vortexed for 5 seconds. This mixture can then be left at room temperature until aIl the samples have been extracted (> 1 hour). The extracts are centrifuged at 13,000 rpm for 1 minute and 300 µl of the supernatant transferred to a fresh Eppendorf tube. This supernatant is mixed with 300 µl propane and left at room temperature for 2 minutes. Following centrifugation at 13,000 rpm for 5 minutes, the pellet is vacuum dried and dissolved in 100 µl 1 X TE. This DNA is stable at 4°C for greater than one year. 2,5 µl of this sample is sufficient for standard 50 µI PCR (Figure 1). When older tissue is used this may be increased to 25 µl without any deleterious effect on the PCR. Using this protocol we have found it possible te process hundreds of individual samples in a single working day. In order te demonstrate one application of the method, genomic DNA was prepared from five Brassica napus plants putatively transformed with the béta- glucuronidase (GUS) reporter gene construct PJIT119 (2. 3). PCR was then performed with 35S promoter and GUS gene specific primers in a Techne PH using the following conditions: 95°C for 45 seconds, 66°C 30 seconds, 73°C fer 2,5 minutes, for 35 cycles. One hal the, PCR reaction was then analysed on a 1,5% agarose gel visualised by ethidium bromide staining (see Figure I). In on to confirm that te observed PCR products correspond te presence of the GUS gene construct, the individual plants were also subjected to a standard GUS enzyme assay (2). The plants in lanes 4, 5, 7 and 8 where shown te have high levels cf GUS enzyme activity whereas no GUS enzyme activity could be detected in plants 3 and C (Data not shown), confirming the results obtained with the PCR analysis.
REFERENCES
1. Saiki, R.K. et al. (1988) Science 239, 487-491.
2. Jefferson, R.A. (1987) Plant Mol. Biol. Rep 5. 387-405
3. Gerineau F. et al. (1990) Plant Mol. Biol. 15. 127-136.
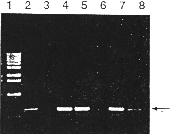
Figure 1. Separation of PCR products following amplification. Lane 1 containts BRL 1 kb molecular weigtht markers. Lane 2 Contains PCR product from 1 pJ1T119 p1asmid DNA (positive control). Lane 3 contains PCR product from an untransformed plant. Lanes 4 - 8 contain PCR products from individual putatively transformed plants. The expected PCR product (580bp) is indicated with an arrow.